Une boussole "moléculaire" montre la voie vers la réduction de l'expérimentation animale
Des scientifiques* développent un outil logiciel intelligent pour évaluer les risques chimiques
Ces dernières années, les méthodes d'apprentissage automatique sont devenues de plus en plus importantes pour l'évaluation des risques des composés chimiques. Mais elles sont aussi une "boîte noire" en raison du manque de traçabilité et de transparence, ce qui entraîne le scepticisme des experts et des autorités d'autorisation. Afin d'augmenter la confiance dans ces modèles, des chercheurs* de l'Université de Vienne ont identifié les domaines dans lesquels ces modèles présentent des faiblesses. Ils ont développé à cet effet un outil logiciel innovant ("MolCompass").
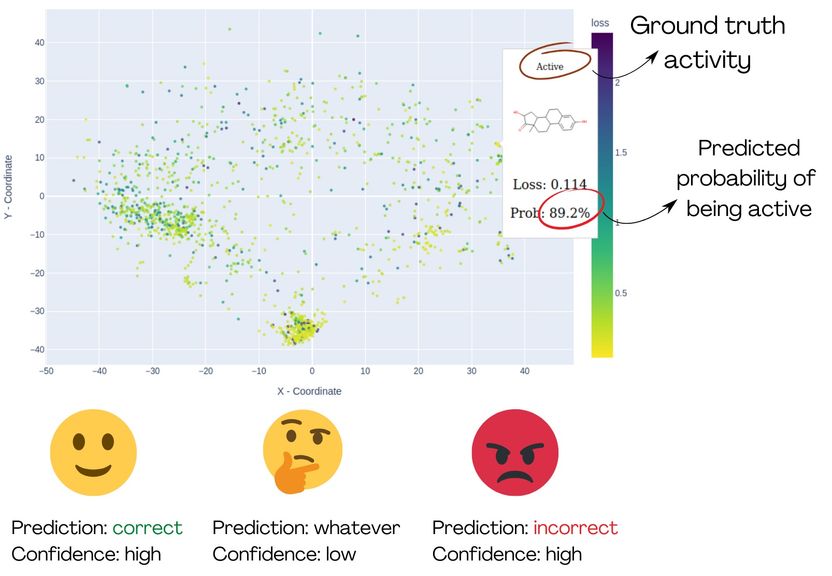
La démonstration de MolCompass illustre comment les toxicologues* informatiques peuvent identifier les régions concernées de l'espace chimique. À l'aide du logiciel, les toxicologues* peuvent repérer les régions où le modèle étudié prédit l'activité de manière erronée avec un haut degré de certitude.
Sergey Sosnin
Pendant de nombreuses décennies, les nouveaux médicaments et produits agrochimiques ont été principalement testés sur les animaux. Ces tests sont coûteux, posent des problèmes éthiques et échouent souvent à prédire avec précision les effets secondaires chez l'homme. Dans le cadre du projet RISK-HUNT3R, soutenu par l'Union européenne, des chercheurs de l'Université de Vienne travaillent au développement de la prochaine génération de méthodes d'évaluation des risques de nouvelles substances sans recours aux animaux. Des méthodes assistées par ordinateur permettent désormais d'évaluer entièrement par ordinateur les risques toxicologiques et écologiques de nouveaux produits chimiques, sans qu'il soit nécessaire de synthétiser et de tester les composés chimiques. Mais une question demeure : Quelle est la fiabilité de ces modèles informatiques ?
Il s'agit de faire des prédictions fiables.
Pour étudier ce problème de plus près, Sergey Sosnin, Senior Scientist au sein du groupe de recherche en pharmaco-informatique de l'université de Vienne, s'est concentré sur la classification binaire. Dans ce cas, un modèle d'apprentissage automatique fournit une probabilité de 0 % à 100 % indiquant si un composé chimique est actif ou non (par exemple, toxique ou non toxique, bioaccumulable ou non bioaccumulable, un liant ou non liant à une protéine humaine spécifique). Cette probabilité reflète la confiance du modèle dans sa prédiction. Idéalement, le modèle ne devrait donner des valeurs proches de 0% (inactif certain) ou de 100% (actif certain) que si les prédictions sont correctes. Si le modèle n'est pas sûr et donne un score de confiance de 51 %, par exemple, ces prédictions devraient être rejetées et des méthodes alternatives d'évaluation des risques devraient être utilisées. Un problème survient toutefois lorsque le modèle fournit des prédictions erronées avec des probabilités élevées.
"C'est le véritable scénario cauchemardesque pour les toxicologues*", déclare Sergey Sosnin. "Si un modèle prédit qu'un composé est non toxique avec 99 % de certitude, mais que le composé est en fait toxique, il n'y a aucun moyen de savoir que quelque chose s'est mal passé". La seule solution consiste à identifier à l'avance les zones de l'espace chimique - c'est-à-dire les classes possibles de composés organiques - dans lesquelles le modèle présente des "points aveugles" et à les éviter. Pour ce faire, les chercheurs qui évaluent le modèle doivent vérifier les résultats prédits pour des milliers de composés chimiques, une tâche fastidieuse et sujette à erreurs.
Surmonter cet obstacle important
"Pour aider ces chercheurs", poursuit Sosnin, "nous avons développé des outils graphiques interactifs qui projettent les composés chimiques sur un plan 2D, comme des cartes géographiques. Nous utilisons des couleurs pour mettre en évidence les composés qui ont été prédits avec un haut degré d'erreur, de sorte que les utilisateurs* puissent les identifier comme des groupes de points rouges. La carte est interactive et permet aux utilisateurs d'examiner l'espace chimique et d'explorer les zones préoccupantes".
La méthodologie a été testée sur un modèle de liaison au récepteur des œstrogènes. Après l'analyse visuelle de l'espace chimique, il est apparu clairement que le modèle fonctionnait bien pour les stéroïdes et les biphényles polychlorés, par exemple, mais qu'il échouait complètement pour les petits composés non cycliques et ne devait donc pas être utilisé pour ces derniers.
Le logiciel développé dans le cadre de ce projet est disponible en libre accès pour la communauté scientifique sur GitHub. Sergey Sosnin espère que MolCompass aidera les chimistes* et les toxicologues* à mieux comprendre les limitations des modèles informatiques. Cette étude est un pas en avant vers un avenir où les tests sur les animaux ne seront plus nécessaires et où le seul poste de travail des toxicologues* sera un bureau avec un ordinateur.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Allemand peut être trouvé ici.
Publication originale
Autres actualités du département science