Une méthode de calcul fiable et efficace pour trouver les états de transition dans les réactions chimiques
Annonces



Une méthode de calcul pour trouver les états de transition dans les réactions chimiques, réduisant considérablement les coûts de calcul avec une grande fiabilité, a été conçue. Par rapport à la méthode existante la plus répandue, la méthode actuelle réduit le coût total de calcul d'environ 50 à 70 %. Ce développement, disponible sur GitHub, devrait accélérer les progrès dans la science des matériaux, en rendant l'exploration des réactions chimiques plus accessible et plus efficace. Cela pourrait conduire à des découvertes scientifiques et à des innovations technologiques plus rapides.
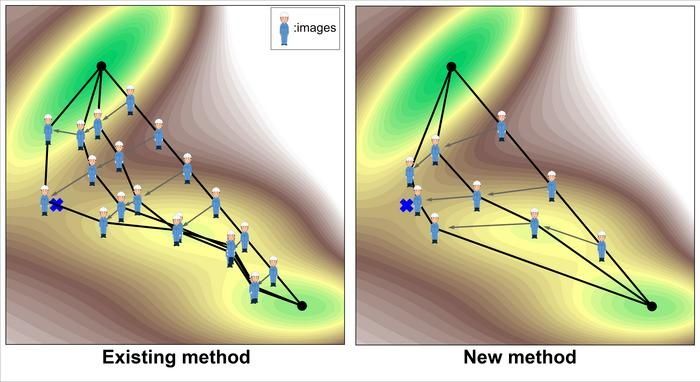
ILLUSTRATION CONCEPTUELLE DE LA RECHERCHE D'UN ÉTAT DE TRANSITION. LES POINTS SITUÉS LE LONG DU CHEMIN DE RÉACTION (SOUVENT APPELÉS IMAGES, MÉTAPHORIQUEMENT REPRÉSENTÉS PAR UNE PERSONNE) SONT MIS À JOUR DE MANIÈRE INCRÉMENTIELLE POUR TROUVER UN ÉTAT DE TRANSITION (MARQUE EN FORME DE CROIX).
Shin-ichi Koda
Dans les réactions chimiques, les substances passent d'un état énergétiquement stable à un autre, en passant par un état de transition instable. Ce processus s'apparente à la recherche de l'itinéraire le moins élevé pour traverser une montagne d'un côté à l'autre. La compréhension de l'état de transition - le sommet de ce chemin de montagne métaphorique - est cruciale pour une compréhension approfondie des mécanismes de réaction. Cependant, en raison de la nature transitoire et instable de ces états, leur observation et leur identification expérimentales sont difficiles et nécessitent souvent une exploration informatique.
Cette étude se concentre sur les méthodes informatiques permettant de trouver un état de transition entre un réactif et un produit connus. Ce type de recherche d'état de transition optimise le chemin reliant le produit et le réactif de manière à ce qu'il passe par l'état de transition. Étant donné que le chemin est généralement représenté par de multiples points sur le chemin (souvent appelés images, métaphoriquement représentées par des personnes dans la figure 1), le chemin est en fait optimisé par une mise à jour incrémentale des images.

La méthode la plus couramment utilisée aujourd'hui est la méthode de la bande élastique bousculée (NEB) (figure 1, à gauche). L'une des principales difficultés de cette méthode est qu'elle est coûteuse en termes de calcul. Il y a deux raisons principales à cela. La première est qu'elle nécessite un grand nombre d'images pour augmenter la résolution de la recherche. L'autre raison est que le principe de recherche n'est pas variationnel (c'est-à-dire qu'il minimise une fonction objective), de sorte que le nombre de mises à jour par image tend également à être élevé.
La nouvelle méthode mise en œuvre dans cette étude résout ces problèmes de manière innovante (figure 1, à droite). Tout d'abord, le nombre d'images peut être réduit à environ 3, puisque seule la région autour de l'état de transition fait l'objet d'une recherche intensive. En outre, le principe de recherche est variationnel et peut donc être résolu plus efficacement. Plus précisément, la fonction objective est définie comme l'intégrale linéaire de l'exponentielle de l'énergie le long de la trajectoire.
La performance de notre nouvelle méthode a été évaluée sur 121 réactions chimiques et les résultats ont été comparés à la méthode NEB et à sa version améliorée. Tout d'abord, la présente méthode a correctement identifié les états de transition dans 98% des cas. Cette précision est bien supérieure à celle de la méthode NEB et comparable à celle de la version améliorée. Deuxièmement, la présente méthode a montré une réduction significative du coût total de calcul - environ 70% de moins que la méthode NEB et 50% de moins que sa version améliorée.
Pour faciliter une application plus large, nous avons mis notre programme de calcul à disposition sur GitHub (github.com/shin1koda/dmf). Écrit en Python et conçu pour être utilisé avec l'environnement de simulation atomique (ASE), il permet aux chercheurs d'explorer facilement les états de transition en spécifiant les réactifs et les produits.
Pour l'avenir, les implications de cette recherche sont vastes. En facilitant et en accélérant la recherche des états de transition, notre méthode est prête à accélérer les recherches et les développements dans tous les domaines des sciences naturelles utilisant la chimie computationnelle.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale

Autres actualités du département science































