Un programme d'apprentissage automatique pour les jeux inspire le développement d'un outil scientifique révolutionnaire
Un nouvel outil d'IA modélise en un temps record le comportement de clusters de nanoparticules
Nous apprenons de nouvelles compétences par la répétition et l'apprentissage par renforcement. Par essais et erreurs, nous répétons des actions qui donnent de bons résultats, nous essayons d'éviter les mauvais résultats et nous cherchons à améliorer ceux qui se situent entre les deux. Les chercheurs conçoivent actuellement des algorithmes basés sur une forme d'intelligence artificielle qui utilise l'apprentissage par renforcement. Ils les appliquent pour automatiser la synthèse chimique, la découverte de médicaments et même des jeux comme les échecs et le go.
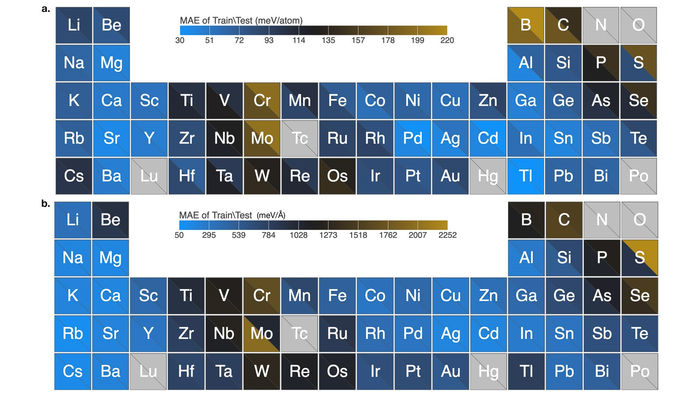
Le graphique montre l'excellente performance de l'algorithme pour les prédictions de champ de force de nanoclusters élémentaires couvrant 54 éléments du tableau périodique. Les données montrent une erreur absolue moyenne très faible.
Image by Argonne National Laboratory
Des scientifiques du laboratoire national Argonne du ministère américain de l'énergie (DOE) ont mis au point un algorithme d'apprentissage par renforcement pour une autre application encore. Il sert à modéliser les propriétés des matériaux à l'échelle atomique et moléculaire et devrait accélérer considérablement la découverte de matériaux.
Comme les humains, cet algorithme "apprend" à résoudre des problèmes à partir de ses erreurs et de ses réussites. Mais il le fait sans intervention humaine.
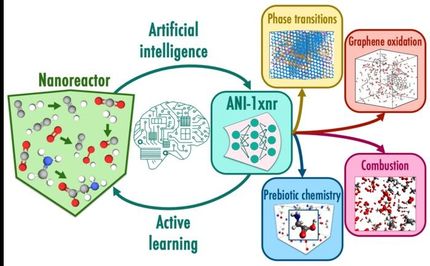
Historiquement, l'Argonne a été un leader mondial dans la modélisation moléculaire. Il s'agit de calculer les forces entre les atomes d'un matériau et d'utiliser ces données pour simuler son comportement dans différentes conditions au fil du temps.
Toutefois, les modèles antérieurs reposaient en grande partie sur l'intuition et l'expertise humaines et nécessitaient souvent des années d'efforts laborieux. L'algorithme d'apprentissage par renforcement de l'équipe réduit ce temps à quelques jours ou quelques heures. Il permet également d'obtenir des données de meilleure qualité que ce que permettent les méthodes conventionnelles.
"Notre inspiration a été AlphaGo", a déclaré Sukriti Manna, assistante de recherche au Center for Nanoscale Materials (CNM) d'Argonne, une installation utilisatrice du DOE Office of Science. "C'est le premier programme informatique à avoir battu un champion du monde de Go".
Le plateau standard du jeu de go compte 361 cases positionnées, soit beaucoup plus que les 64 cases d'un échiquier. Cela se traduit par un très grand nombre de configurations possibles du plateau. La clé pour qu'AlphaGo devienne un champion du monde a été sa capacité à améliorer ses compétences grâce à l'apprentissage par renforcement.
L'automatisation de la modélisation moléculaire est, bien sûr, très différente d'un programme informatique de Go. "L'un des défis auxquels nous avons été confrontés est similaire au développement de l'algorithme requis pour les voitures à conduite autonome", a déclaré Subramanian Sankaranarayanan, chef de groupe au CNM d'Argonne et professeur associé à l'université de l'Illinois à Chicago.
Alors que le panneau Go est statique, les environnements de circulation changent continuellement. La voiture autonome doit interagir avec d'autres voitures, varier les itinéraires, les panneaux de signalisation, les piétons, les intersections, etc. Les paramètres liés à la prise de décision évoluent constamment dans le temps.
La résolution de problèmes difficiles du monde réel dans le domaine de la découverte et de la conception de matériaux implique également une prise de décision continue dans la recherche de solutions optimales. L'algorithme de l'équipe comprend des arbres de décision qui distribuent un renforcement positif en fonction du degré de réussite de l'optimisation des paramètres du modèle. Le résultat est un modèle capable de calculer avec précision les propriétés des matériaux et leurs variations dans le temps.
L'équipe a testé avec succès son algorithme avec 54 éléments du tableau périodique. Leur algorithme a appris à calculer les champs de force de milliers d'amas nanométriques pour chaque élément et a effectué les calculs en un temps record. Ces nanoclusters sont connus pour leur chimie complexe et la difficulté qu'ont les méthodes traditionnelles à les modéliser avec précision.
"Cela revient à effectuer les calculs de plusieurs thèses de doctorat en quelques jours, au lieu de plusieurs années", a déclaré Rohit Batra, expert du CNM en matière d'outils d'apprentissage automatique et de données. L'équipe a effectué ces calculs non seulement pour les nanoclusters d'un seul élément, mais aussi pour les alliages de deux éléments.
"Notre travail représente une avancée majeure dans ce type de développement de modèles pour la science des matériaux", a déclaré Sankaranarayanan. "La qualité de nos calculs pour les 54 éléments avec l'algorithme est bien supérieure à l'état de l'art."
L'exécution de l'algorithme de l'équipe a nécessité des calculs avec de grands ensembles de données sur des ordinateurs à haute performance. À cette fin, l'équipe a fait appel au cluster d'ordinateurs Carbon du CNM et au superordinateur Theta de l'Argonne Leadership Computing Facility, un centre d'utilisateurs du DOE Office of Science. Elle a également fait appel aux ressources informatiques du National Energy Research Scientific Computing Center, une installation du DOE Office of Science située au Lawrence Berkeley National Laboratory.
"L'algorithme devrait accélérer considérablement le temps nécessaire pour relever les grands défis dans de nombreux domaines de la science des matériaux", a déclaré Troy Loeffler, chimiste théoricien et informaticien au CNM. Les exemples incluent les matériaux pour les dispositifs électroniques, les catalyseurs pour les processus industriels et les composants de batteries.
Note: Cet article a été traduit à l'aide d'un système informatique sans intervention humaine. LUMITOS propose ces traductions automatiques pour présenter un plus large éventail d'actualités. Comme cet article a été traduit avec traduction automatique, il est possible qu'il contienne des erreurs de vocabulaire, de syntaxe ou de grammaire. L'article original dans Anglais peut être trouvé ici.
Publication originale

Autres actualités du département science