Catalyst Research: Molecular Probes Require Highly Precise Calculations
Scientists Use Advanced Methods with Hybrid Functionals for Analysis of Active Sites
catalysts are indispensable for many technologies. To further improve heterogeneous catalysts, it is required to analyze the complex processes on their surfaces, where the active sites are located. Scientists of Karlsruhe Institute of Technology (KIT), together with colleagues from Spain and Argentina, have now reached decisive progress: As reported in Physical Review Letters, they use calculation methods with so-called hybrid functionals for the reliable interpretation of experimental data.
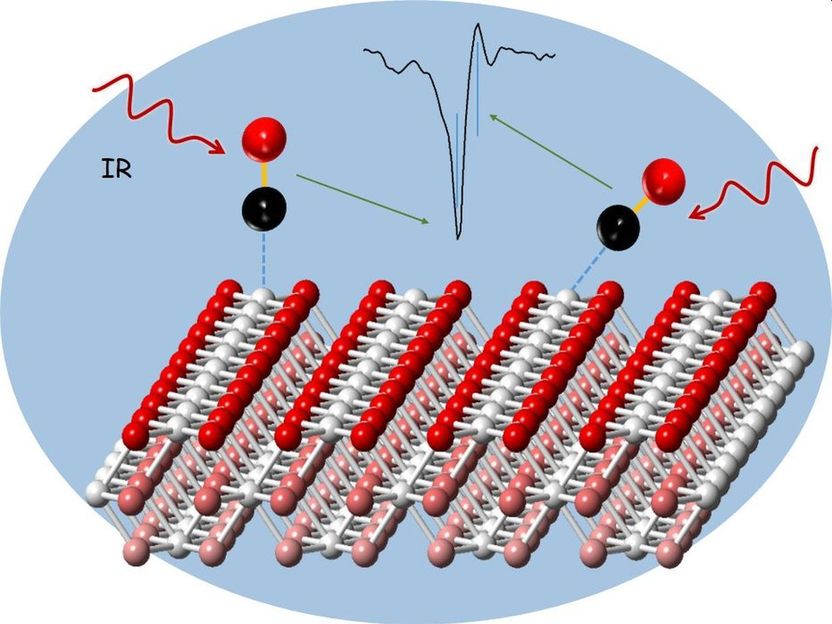
Analysis of a cerium oxide catalyst using carbon monoxide probe molecules and infra-red reflection absorption spectroscopy.
IFG/KIT
Many important technologies, such as processes for energy conversion, emission reduction, or the production of chemicals, work with suitable catalysts only. For this reason, highly efficient materials for heterogeneous catalysis are gaining importance. In heterogeneous catalysis, the material acting as a catalyst and the reacting substances exist in different phases as a solid or gas, for instance. Material compositions can be determined reliably by various methods. Processes taking place on the catalyst surface, however, can be detected by hardly any analysis method. “But it is these highly complex chemical processes on the outermost surface of the catalyst that are of decisive importance,” says Professor Christof Wöll, Head of KIT’s Institute of Functional Interfaces (IFG). “There, the active sites are located, where the catalyzed reaction takes place.”
Precise Examination of the Surface of Powder Catalysts
Among the most important heterogeneous catalysts are cerium oxides, i.e. compounds of the rare-earth metal cerium with oxygen. They exist in powder form and consist of nanoparticles of controlled structure. The shape of the nanoparticles considerably influences the reactivity of the catalyst. To study the processes on the surface of such powder catalysts, researchers recently started to use probe molecules, such as carbon monoxide molecules, that bind to the nanoparticles. These probes are then measured by infrared reflection absorption spectroscopy (IRRAS). Infrared radiation causes molecules to vibrate. From the vibration frequencies of the probe molecules, detailed information can be obtained on the type and composition of the catalytic sites. So far, however, interpretation of the experimental IRRAS data has been very difficult, because technologically relevant powder catalysts have many vibration bands, whose exact allocation is challenging. Theoretical calculations were of no help, because the deviation from the experiment, also in the case of model systems, was so large that experimentally observed vibration bands could not be allocated precisely.
Long Calculation Time – High Accuracy
Researchers of KIT’s Institute of Functional Interfaces (IFG) and Institute of Catalysis Research and Technology (IKFT), in cooperation with colleagues from Spain and Argentina coordinated by Dr. M. Verónica Ganduglia-Pirovano from Consejo Superior de Investigaciones Científicas (CSIC) in Madrid, have now identified and solved a major problem of theoretical analysis. As reported in Physical Review Letters, systematic theoretical studies and validation of the results using model systems revealed that theoretical methods used so far have some fundamental weaknesses. In general, such weaknesses can be observed in calculations using the density functional theory (DFT), a method with which the quantum mechanics basic state of a multi-electron system can be determined based on the density of the electrons. The researchers found that the weaknesses can be overcome with so-called hybrid functionals that combine DFT with the Hartree-Fock method, an approximation method in quantum chemistry. This makes the calculations very complex, but also highly precise. “The calculation times required by these new methods are longer by a factor of 100 than for conventional methods,” says Christof Wöll. “But this drawback is more than compensated by the excellent agreement with the experimental systems.” Using nanoscaled cerium oxide catalysts, the researchers demonstrated this progress that may contribute to making heterogeneous catalysts more effective and durable.
Original publication

Other news from the department science




























































