Metallic surfaces help molecular quantum switch
Quantum molecular switches operate by hydrogen tunneling with some help
Advertisement



The quantum dynamics of hydrogen is central to many problems in nature, being strongly influenced by the environment in which it takes place. In their contribution to PRL, members of the Lise Meitner Group at the MPSD address hydrogen transfer within a supported molecular switch, showing that the surface support can play a decisive role in the tunneling reaction.
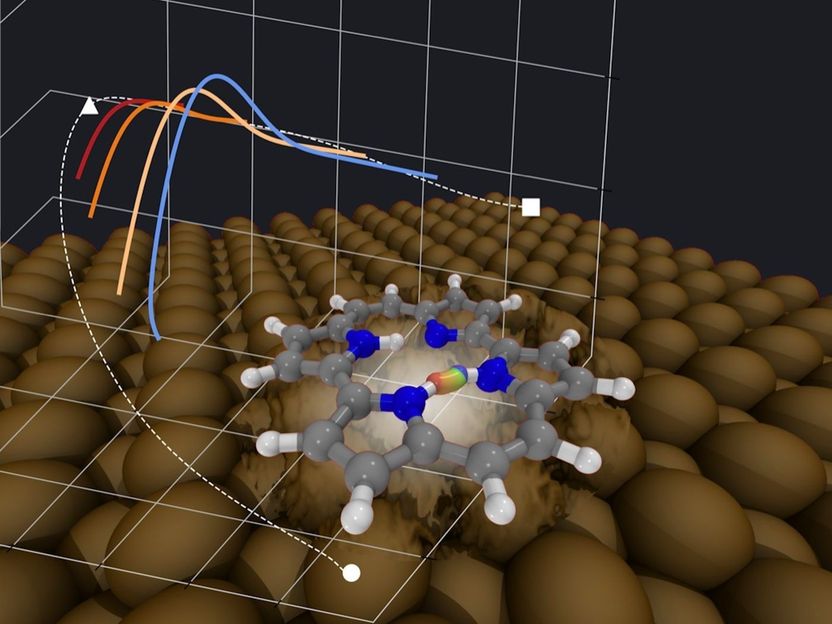
Within the porphycene molecule adsorbed on metallic fcc[110] surfaces, hydrogen transfer reactions happen through nuclear tunneling even just below room temperature. The figure schematically shows an instanton tunneling path, obtained on a full-dimensional first-principles potential energy surface, in real space and projected on selected molecular coordinates. The fluctuation of surface atoms can increase the tunneling rate by a few orders of magnitude.
© Mariana Rossi
In the constant quest to miniaturize technological components, molecule-based nanotechnology becomes an ever-growing field of interest. In this field, single molecules become the fundamental components of electronic devices. The sheer variety of possible molecular architectures and the possibility of precisely manipulating molecular synthesis opens the door to endless functional components. However, the key challenge is to gain control over these functions in the nanoscale, where quantum mechanical effects become prominent.
The porphycene molecule is one example of a prototype molecular switch. Porphycene is a structural isomer of porphyrin with strong H-bonds in its inner cavity. Its switching capability relies on a very fundamental reaction in chemical physics: a double hydrogen transfer that can swap the positions of the hydrogens in the inner cavity and thus define different states (on/off) of the molecule — a process called tautomerisation. In order to control and measure the atomic structure and the switching rate of these molecular units, they are typically immobilized by placing them in contact with metal surfaces. This situation calls for researchers to understand hydrogen dynamics within an environment that includes qualitatively different types of interaction between atoms within the molecule and between the molecule and the surface.

In this context, porphycene has been extensively studied by single-molecule experimental techniques. Researchers have observed several puzzling aspects of the tautomerisation rate over different temperature ranges, including temperatures where atoms no longer behave like classical particles, but can tunnel through barriers instead. Using a mountain as an analogy, the atoms would travel instantaneously between two valleys in a straight line under the mountain, instead of taking the time to go up and down over it.
In their new work just published in PRL, Yair Litman and Mariana Rossi address this supported molecular switch with state-of-the-art methodology and new computer algorithms: a combination of density-functional theory with ring-polymer instantons. These methods finally allowed such systems to be studied with full-scale atomistic simulations that treat both electrons and nuclei as quantum mechanical particles. The authors show that for porphycene adsorbed on Cu(110) and Ag(110) surfaces, the hydrogen transfer reaction indeed presents a large contribution from nuclear tunneling even at temperatures not far below room temperature.
Quite surprisingly, the authors discovered that with the lowering of the temperature, heavy surface atoms like copper participate in the intramolecular hydrogen tunneling reaction and can cause an increase in the tunneling rate by up to two orders of magnitude at a temperature of around 80 K. The stronger the interaction of the molecule with the surface (hybridisation of electronic orbitals), the more pronounced is the participation of the surface atoms in the tunneling event.
Notably, the authors also explained an unconventional temperature dependence of the tunneling rate, which was previously observed in experiments. It is caused by the existence of an intermediate metastable structure in the reaction, which exists for such a short period of time (~100 picoseconds, with one picosecond being a trillionth of a second) that it could not be detected by the experimental techniques previously employed in this system. Understanding this mechanism, the authors could also explain different temperature dependence regimes of the rate in the tunneling regime and propose a simple model to predict this temperature dependence for this switch adsorbed on other metallic surfaces.
These are important new insights into the fact that certain characteristics of the surface support can influence nuclear quantum mechanical properties of the switching reaction in these, and probably other, molecules. They also demonstrate that single-crystal substrates are an ideal platform where cutting-edge theory and experiment can come together to provide a deeper understanding of nuclear quantum dynamics in complex environments. Such findings are of considerable fundamental importance and can also guide the design and interpretation of experimental architectures in molecular nanotechnology development.
Original publication




Other news from the department science




























































